全球顶赛连续霸榜包揽前二!昇思MindSpore蛋白质结构预测模型超越Alphafold原型
全球顶赛连续霸榜包揽前二!昇思MindSpore蛋白质结构预测模型超越Alphafold原型

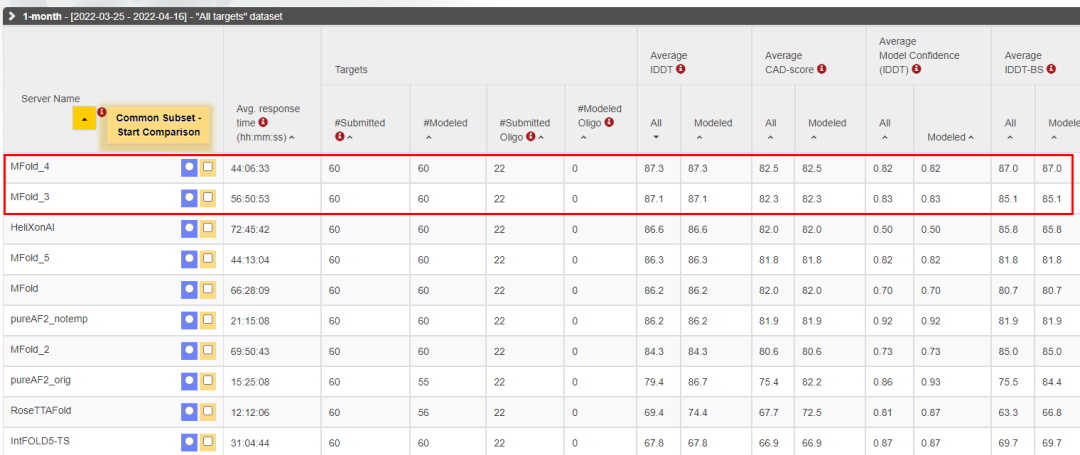
近期,基于全场景AI框架昇思MindSpore 开发的蛋白质结构预测模型在全球持续蛋白质结构预测竞赛CAMEO(Continous Automated Model EvaluatiOn)上连续三周霸榜,并包揽月榜前二,显示出昇腾AI基础软硬件平台在AI for Science领域的强大能力。
(4月21日最新月榜竞赛结果)

由瑞士生物信息研究所和巴塞尔大学联合举办的 CAMEO(Continous Automated Model EvaluatiOn)——全球持续蛋白质结构预测竞赛,被认为是蛋白质结构预测领域最重要的比赛之一,有很多研究者更视其为仅次于CASP的重量级比拼。
CAMEO 竞赛的规则是,每位参赛者需每周对 20 个由世界范围内的结构生物学家最新破解出的蛋白质结构进行预测,其分数和名次每周都会进行在线更新。因此,该竞赛吸引来不少世界范围内生物计算领域的前沿模型“选手”。
CAMEO ( https://www.cameo3d.org/ )作为一个社区项目,其不断应用蛋白质结构预测社区建立的质量评估标准。 由于不同科学应用的准确度要求各不相同,因此没有“一刀切”的分数。 因此,CAMEO 提供了各种分数 - 评估预测的不同方面(覆盖范围、局部准确性、完整性等)以反映这些要求。

PDB(Protein Data Bank,https://www.rcsb.org/)每周发布程序的一部分是在实际发布前五天(即星期五)发布下周发布的条目序列。CAMEO 收集这些序列并在经过一些预处理后将它们提交给注册的服务器。评估可以在 PDB 发布实际结构后进行 - 通常在下周三。
CAMEO 支持的类别是蛋白质结构建模 (3D)、蛋白质模型质量评估 (QE) 以及结构和复合物 (Beta 3D)。蛋白质接触预测 (CP) 和配体结合位点 (LB) 已停止使用。
CAMEO 服务器可以注册为公共服务器,其全名和结果可供所有人使用,也可以注册为开发服务器,其中名称被伪装('serverX')并且所有评分都被执行并且对其他方法开发人员可见,但对公众不可见。请参阅完整注册服务器列表(https://www.cameo3d.org/cameong\_servers/ )。
CAMEO 目标基于每周预发布的新 PDB 结构,这些结构将提交给所有注册的服务器。模板覆盖至少 70% 序列且序列同一性超过 85% 的目标不提交 3D - 蛋白质结构类别。这也适用于序列长度 > 250 且未发现的氨基酸少于 40 个的靶标。
在选定的时间范围内已评估的提交的单体和同源寡聚目标的总数。实际提交的目标数量可能会更多,因为一些目标将在实验结构审查后被排除在外。
目标的难度是通过评估从服务器接收到的模型的平均准确度 (LDDT) 来定义的。所有服务器的低 lDDT 清楚地表明一个硬目标。简单的平均 lDDT >= 75,中等的平均 lDDT 介于 50 和 75 之间,困难的平均 lDDT < 50。
低于截止值的原子比例类似于预测中 Cα 原子的百分比,该预测偏离目标不超过不同序列相关叠加的指定距离(以 Å 为单位)截止值。该方法使用 0.5、1、2 和 4 Å 的距离截止值,而叠加是使用 LGA 程序计算的。
由上可以看到,昇思MindSpore取得了超过85分档位的相当优异的成绩,而在CAMEO“寸分寸金”的评价体系下,包揽前二更是绝对实力的体现。
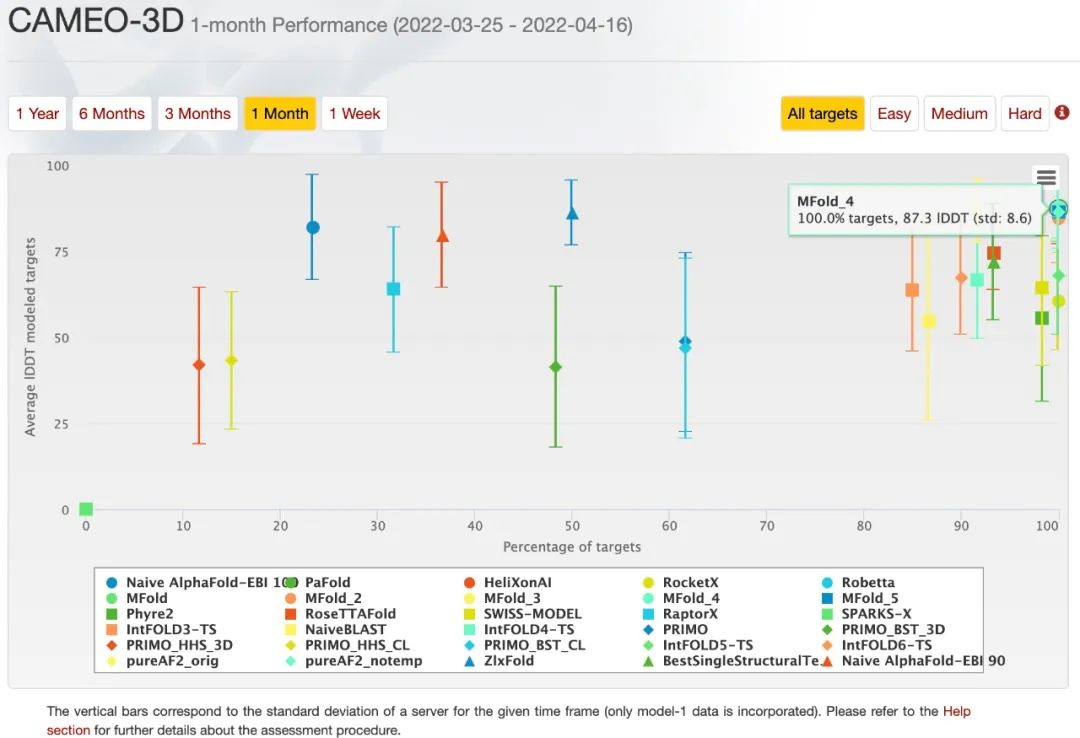

CAD 分数(接触面积差异)提供了一个统一的框架,用于评估具有不同准确性和完整性的单域、多域甚至多亚基蛋白质结构模型。CAD-score 在单域结构上与 GDT-TS 高度相关的同时,更强调模型的物理真实性,并且没有叠加。值范围在 0 到 100 之间(0 差,100 好)。
模型的覆盖率是模型中残基的百分比,其中关于提交序列长度的实验结构中的结构信息可用。一些 CAMEO 靶标的覆盖率很低,因为它们在较大的复合物中没有被解析出蛋白质片段。我们保留将来删除这些目标的权利。
GDT_HA(全局距离测试)分数可识别预测中的残基集,这些残基与目标的偏差不超过不同序列相关叠加的指定 Cα 距离截止值。该方法使用 0.5、1、2 和 4 Å 的距离截止值。帮助部分
GDC 分数识别预测中的残基集,该残基与目标的偏差不超过不同序列相关叠加的指定全原子距离截止值。该方法使用 0.5、1、2 和 4 Å 的距离截止值。(这个分数是用 LGA 计算的,版本 2009/5 “LGA 是一种在蛋白质结构中寻找 3-D 相似性的方法”。
lDDT 分数(所有原子的局部距离差异测试)评估模型的局部原子环境的质量。lDDT 在不同阈值水平下奖励模型中正确预测的原子间距离的比例。lDDT 不依赖于预测和目标结构的全局叠加。
具体来说,将参考蛋白质结构中原子之间的相互作用距离(截止 15 Å)与预测中相应原子之间的距离进行比较。如果两个距离之间的差异在定义的阈值内,则认为交互作用被保留在预测中。最终的 lDDT-all 分数是通过平均以下四个距离差阈值的正确建模交互的分数来计算的:0.5、1、2 和 4 Å(与 GDT_HA 相同的阈值)。基于 Engh 和 Huber 键长和角度的过滤器可消除立体化学违规和空间冲突。CAMEO 还提供基于 Cα 的 lDDT 评分。
lDDT-BS 评分(局部距离差异测试 - 结合位点)测量在目标结构上形成结合位点的残基的准确性。这里的结合位点被定义为参考蛋白质结构中的一组氨基酸残基,在配体的任何原子的 4.0 Å 半径内(离子为 3.0 Å)至少有一个原子。仅考虑与目标形成非共价相互作用的配体。基于黑名单方法,常见的溶剂分子也被排除在分析之外。LDDT 分数仅针对结合位点内的接触计算,自定义包含半径 Ro 为 10 Å,标准阈值为 0.5 Å、1 Å、2 Å 和 4 Å。
当参考结构包含几个生物学相关的配体时,lDDT-BS 得分是各个结合位点的 lDDT-BS 得分的平均值。该分数仅针对实验结构包含配体的目标计算。当结合位点位于同源寡聚结构的界面并且预测未显示相同的寡聚状态时,将评估所有可能的链映射并保留得分最高的映射。不评估仅覆盖部分异质寡聚复合物的目标,因为建模形式可能与其自然状态不同,并且在进化相关的异质寡聚复合物中存在差异。
模型置信度 LDDT 值的评估使用 ROC AUC 分析评估以 Å 为单位的偏差给出的误差估计。如果残留物的局部 lDDT 值高于或等于 0.6,则残留物被分类为正确建模。各个模型页面上显示的局部分数是模型在给定残基位置与参考结构拟合程度的度量。在计算平均准确度时,从输出中提取局部分数。请注意,ROC AUC 没有为太好或太差的模型定义(所有 lDDT 值大于或小于 0.6)。

2021年11月,昇思MindSpore团队与昌平实验室、北京大学生物医学前沿创新中心(BIOPIC)和化学与分子工程学院、深圳湾实验室高毅勤教授课题组联合推出基于AlphaFold2算法的蛋白质结构预测推理工具,并在2022年2月实现训练的全流程打通,效率同比提升2-3倍。
采用昇腾AI基础软硬件平台后,在混合精度下,单步迭代时间由20秒缩短到12秒,性能提升超过60%。依托昇思MindSpore内存复用能力, 训练序列长度由384提升至512。
在训练精度接近AlphaFold2的基础上,昇思MindSpore在算法、规模和软硬件支持等方向上持续改进,本次发布的蛋白质结构预测模型成绩持续刷新业界记录、拿下CAMEO竞赛第一,毫无疑问是昇思MindSpore在蛋白质结构预测领域的又一里程碑,在填补国产AI基础软硬件在蛋白质结构预测领域成绩空白的同时,更加证明了其价值与优越性。
未来,针对蛋白质结构预测问题,昇思MindSpore将携手学术科研界更多合作伙伴,在蛋白质结构预测领域共同探索、持续突破,助力国内相关基础研究,尤其是生命各分支学科的进步,推动创新制药行业的发展。


MindSpore官方资料
官方QQ群 : 486831414
Gitee : https : //gitee.com/mindspore/mindspore