Idea Sharing | DiffDock: A Diffusion Generative Model to Enhance Molecular Docking Accuracy
Idea Sharing | DiffDock: A Diffusion Generative Model to Enhance Molecular Docking Accuracy
Background
For a considerable period, molecular docking was viewed primarily as a search task. Traditional methods typically employed lattice point calculations and fragment growth for spatial recognition between molecules, while energy computations were performed using simulated annealing and genetic algorithms. However, recent advancements in deep learning have reframed molecular docking as a regression task, leading to an increase in the speed of molecular docking, but without significant improvements in accuracy.
In this blog, I'm excited to discuss the innovative work of Professors Regina Barzilay and Tommi Jaakkola from MIT CSAIL. They regard molecular docking as a generative task and have developed a generative diffusion model (DGM). Their DiffDock model improves the docking speed by 3 to 12 times and achieves a success rate of 38% in docking tasks, significantly outperforming the previous state-of-the-art deep learning models (20%) and conventional space-based docking (23%) methods. Moreover, previous methods obtain a maximum of 10.4% success rate for folded complex structure docking, while DiffDock can reach 21.7%.
Paper:
DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking
Link:
Code:
1. Model
The following figure shows the basic structure of DiffDock, which takes the separate ligand and protein as inputs. Randomly sampled initial poses are denoised via a reverse diffusion over translational, rotational, and torsional degrees of freedom. The sampled poses are ranked by the confidence model to produce a final predictions and confidence scores.
Figure 1. Overview of DiffDock
1.1 Ligand Pose
A ligand pose refers to the location of all atoms of a ligand in a 3D space. In principle, we can regard a pose X as a point in the 3_n_ manifold, where n is the number of atoms.
However, this encompasses less degree of freedom than 3_n_. That is because bond lengths, angles, and small rings in the ligand are basically rigid, so that the ligand flexibility lies almost entirely in the torsion angles at rotatable bonds. Conventional docking methods and most machine learning methods take a seed transformation c of the separate ligand in the 3_n_ manifold as the input, and change only the relative position and the torsion degrees of freedom in the final bound conformation. The space of ligand poses consistent with c is, therefore, an (m + 6)-dimensional submanifold, where m represents the number of rotatable bonds and the value 6 indicates the rototranslations relative to the fixed protein.
For a manifold, any ligand pose with a seed conformation can be reached by a combination of translations, rotations, and changes to torsion angles. Following this paradigm, the authors take the seed conformation c as input and formulate molecular docking as a task of learning the probability distribution p_c(x | y), conditioned on the protein structure y. Moreover, they define a one-to-one mapping (transformation) from one manifold to another "better" manifold, where the diffusion kernel can be directly sampled.
1.2 Diffusion Model
For the foregoing three transformations (translations, rotations, and changes to torsion angles) of the ligand pose, the random diffusion equation can be defined in the following form:
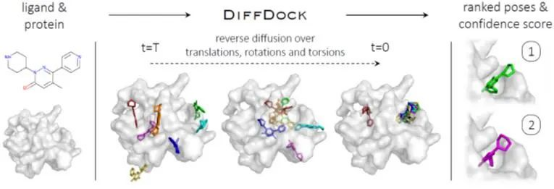
Figure 1. Overview of DiffDock
1.1 Ligand Pose
A ligand pose refers to the location of all atoms of a ligand in a 3D space. In principle, we can regard a pose X as a point in the 3_n_ manifold, where n is the number of atoms.
However, this encompasses less degree of freedom than 3_n_. That is because bond lengths, angles, and small rings in the ligand are basically rigid, so that the ligand flexibility lies almost entirely in the torsion angles at rotatable bonds. Conventional docking methods and most machine learning methods take a seed transformation c of the separate ligand in the 3_n_ manifold as the input, and change only the relative position and the torsion degrees of freedom in the final bound conformation. The space of ligand poses consistent with c is, therefore, an (m + 6)-dimensional submanifold, where m represents the number of rotatable bonds and the value 6 indicates the rototranslations relative to the fixed protein.
For a manifold, any ligand pose with a seed conformation can be reached by a combination of translations, rotations, and changes to torsion angles. Following this paradigm, the authors take the seed conformation c as input and formulate molecular docking as a task of learning the probability distribution p_c(x | y), conditioned on the protein structure y. Moreover, they define a one-to-one mapping (transformation) from one manifold to another "better" manifold, where the diffusion kernel can be directly sampled.
1.2 Diffusion Model
For the foregoing three transformations (translations, rotations, and changes to torsion angles) of the ligand pose, the random diffusion equation can be defined in the following form:
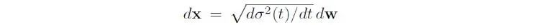
Where x is the pose, w is the Brownian motion, and σ__2 is the noise variance. Diffusion in the case of translations is the simplest, which is a standard Gaussian distribution, while diffusion in the case of rotations and changes to torsion angles is slightly complex (see Nikolayev & Savyolov, 1970[1] and Leach et al., 2022_[2]_).
Although the authors define the diffusion kernel in the (m + 6)-dimensional submanifold, the training and inference process of the diffusion kernel is still performed in 3D coordinates. This is because a full molecular 3D structure, rather than an abstract element in the manifold, is provided to the score model, allowing the model to reason about physical interactions between molecules. This will not be affected by arbitrary definitions of torsion angles and better generalizes to unseen complexes.
1.3 Confidence Model
To obtain training data of the confidence model d(x, y), the authors first run the trained diffusion model to obtain a set of candidate poses for each training sample and generate T/F labels to determine whether RMSD of each candidate pose is below 2Å. The confidence model is then trained with cross-entropy loss, with the goal of predicting an accurate T/F label for each pose. In the inference process, the diffusion model runs to generate N poses in parallel, which then are passed to the confidence model. Finally, the confidence model sorts these poses based on the obtained confidence values with RMSD less than 2Å.
2. Results
The authors use complexes on PDBBind, a dataset of protein-ligand structures collected from PDB, to evaluate the effect of DiffDock generating 10 samples and 40 samples. The results are compared with state-of-the-art search-based methods SMINA, QuickVina-W, GLIDE, GNINA, and Autodock Vina, as well as recent deep learning-based methods EquiBind and TANKBind.
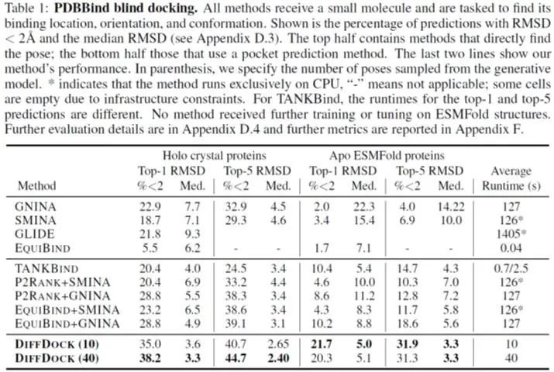
Figure 2. Comparison of DiffDock and other molecular docking methods on PDBBind
The authors also quantify the change of DiffDock performance with the number of generated samples and the accuracy of selecting the best docking result.
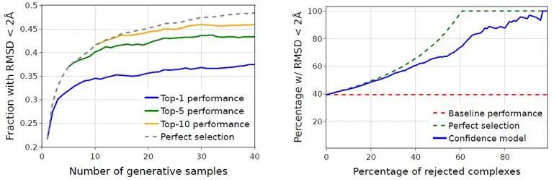
Figure 3. DiffDock performance with the number of generated samples (left) and the accuracy of selecting the best docking result (right)
3. Conclusion
The main contributions of this project are as follows:
● Define the molecular docking task as a generative task.
● Construct a new diffusion process for ligand poses according to the degree of freedom of molecular docking.
● Achieve a 38% top-1 success rate on PDBBind, significantly outperforming the previous state-of-the-art conventional docking (23%) and deep learning (20%) methods.
In general, taking molecular docking as a generative task is very novel. Although DiffDock has improved accuracy compared with some previous methods, it still cannot completely replace conventional methods. It's expected that more new ideas with AI methods would emerge in this field to further promote the development of drug discovery tools.
References
[1] Dmitry I Nikolayev and Tatjana I Savyolov. Normal distribution on the rotation group so (3). Textures and Microstructures, 29, 1970.
[2] Adam Leach, Sebastian M Schmon, Matteo T Degiacomi, and Chris G Willcocks. Denoising diffusion probabilistic models on so (3) for rotational alignment. In ICLR 2022 Workshop on Geometrical and Topological Representation Learning, 2022