Idea Sharing: Allegro Will Be Added to MindSpore Chemistry to Improve the Accuracy of Molecular Potential Energy Prediction
Idea Sharing: Allegro Will Be Added to MindSpore Chemistry to Improve the Accuracy of Molecular Potential Energy Prediction
Background
Molecular modeling, also called molecular simulation, uses theoretical methods and computing technologies to simulate the appearance or properties of chemical molecules. It is the core pillar of computational chemistry, material science, and biology [1].
An ideal method of molecular modeling is quantum chemistry, including ab initio, density functional theory, etc. Although this method considers quantum characteristics inside an atom and has high accuracy, the computing cost is very high and can only be used in relatively small systems. Modeling in a larger scale usually employs molecular dynamics. This method needs to predict the potential energy and atomic forces accurately and effectively, so that the evolution of a complex system over a long time scale can be precisely described. The classical force field method uses only a simple function on atomic coordinates to predict molecular potential energy, which is not accurate, while machine learning interatomic potentials (MLIPs) are expected to offer accurate predictions with high computing efficiency. Among them, the neural network models based on message passing are particularly accurate.
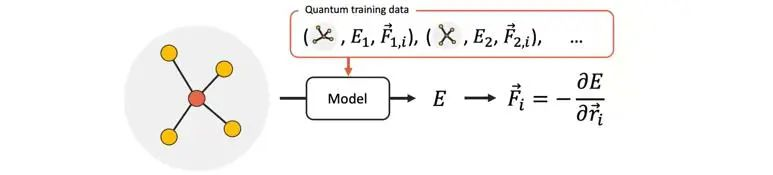
Figure 1 Molecular potential energy prediction using a neural network model
Message passing is a concept in graph neural network (GNNs), where neighboring nodes or edges exchange information and influence embedding update of each other. Message passing consists of three steps (as shown in Figure 2) [2]:
(1) For each node in the graph, gather all the neighboring node embeddings (or messages).
(2) Aggregate all messages via an aggregate function, such as sum.
(3) All aggregated messages are passed through an update function, usually a neural network, to update the target nodes or edges.
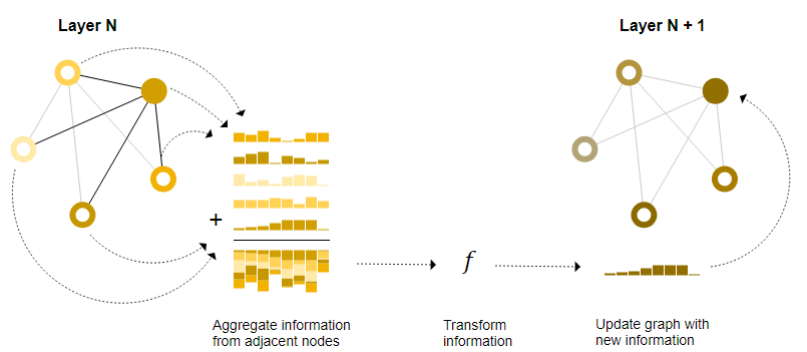
Figure 2 Message passing in a GNN
After multiple times of message passing, a local attribute in the initial graph can affect a remote node. Such a phenomenon is also called convolutions on graphs. Based on the new graph formed after multiple times of message passing, prediction tasks can be implemented [3].
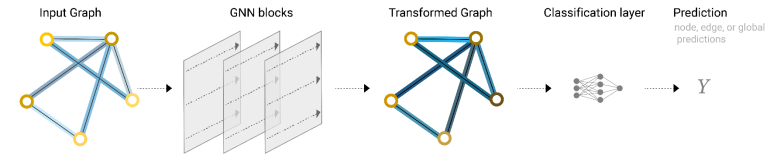
Figure 3 Complete working process of GNNs
In molecular potential energy prediction, geometric GNNs use geometric information (such as distance, angle, and dihedral angle) of molecules and maintain symmetry of physical quantities during computing processes (translation, rotation, and inversion), which is also developing rapidly [4].
Among geometric GNNs, equivariant networks have gained great attention. Taking NequIP [5] as an example, the introduction of equivariance greatly reduces the need for large datasets.
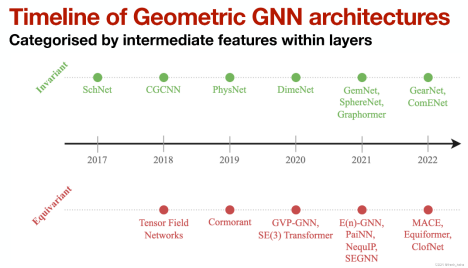
Figure 4 Development of Geometric GNNs
1. Allegro Network Structure
Allegro [6] is an equivariant neural network. For concepts related to equivariance, such as irreducible representations (irreps) and tensor product, see the previous article Idea Sharing: Equivariant Neural Network and e3nn Math Library.
Compared with the traditional approaches based on message passing, Allegro has two main differences: using ordered atom pairs to mark hidden layer features and using a scalar and tensor dual-path architecture instead of the message passing mechanism. Figure 5 shows the overall network architecture.
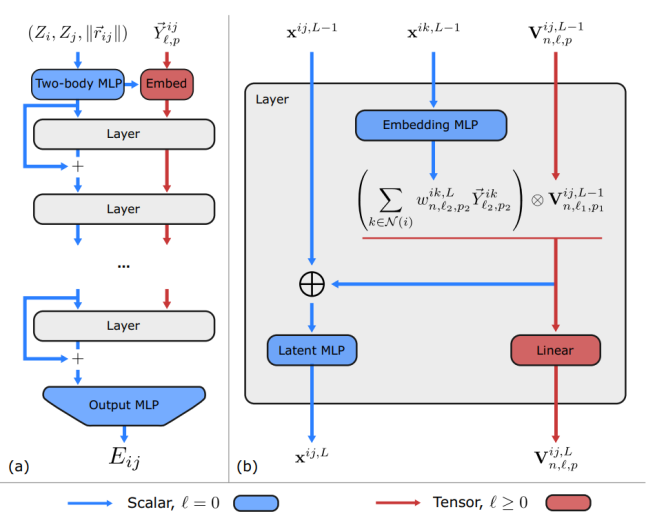
Figure 5 Allegro network architecture
1.1 Hidden Layer Features Marked by Ordered Atom Pairs
The hidden layer features in Allegro are marked by ordered atomic pairs (directed edges) (i,j), where i represents a central atom. Information updating is also completed based on the tensor product of edges.
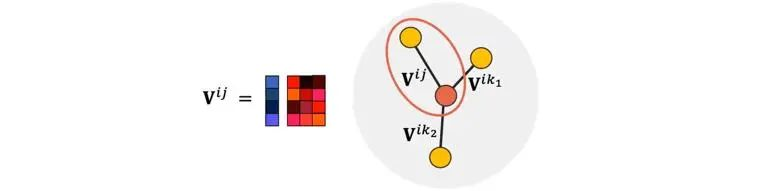
Figure 6 Ordered atom pairs in Allegro
1.2 Scalar and Tensor Dual-Path Architecture
In Figure 5, the blue paths represent the invariant hidden layer space, and x^{ij} represents the hidden layer features (the scalar part, l=0) that maintains invariance. Any operation may be performed on this part. The red paths represent the equivariant hidden layer space, and V^{ij} represents the hidden layer features (the tensor part, l>0) that maintains equivariance. Operations on this part of features need to comply with the equivariance. The initial scalar features x^{ij} of the hidden layers are formed by concatenating the radial basis projection of the atom species and the interatomic distance.
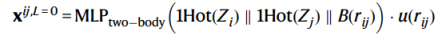
Formula 1
The initial hidden tensor features V^{ij} is the linear projections of the spherical harmonics of the distance vectors from the central atoms to the neighbor atoms.
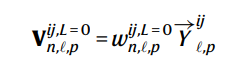
Formula 2
Compared with scalar operations, tensor operations in the equivariant space consume more computing power. The purpose of the dual-path design is to obtain the information of equivariant features with a small computation cost.
Specifically, each layer includes four parts:
(1) An embedding MLP that generates weights to embed the neighboring environment of the central atom to be used as the tensor product of each edge pairs
(2) A tensor product that performs weighted sum in the central atom dimension
(3) A latent MLP to update x^{ij} based on the scalar path output (l=0, p=1) of the tensor product (irreps) concatenated to the original hidden layer scalar features
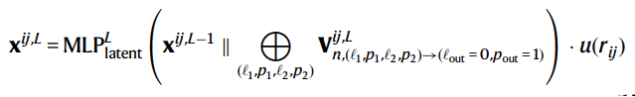
Formula 3
(4) An equivariant linear operation on the tensor product to update V^{ij}
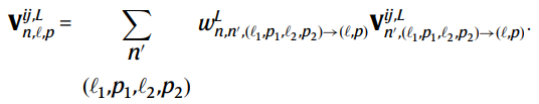
Formula 4
Due to the bilinearity of the tensor product operation, only one tensor product operation needs to be performed for each directed edge of each central atom, as shown in the following formula:
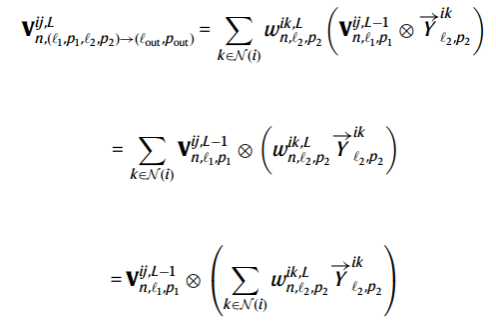
Formula 5
Residuals are also introduced in the scalar space between layers to facilitate information propagation.
1.3 Energy Decomposition
The molecular potential energy of a system can be decomposed into the sum of all atomic energies in the system: E_system = Σ(i)E_i. Allegro further decomposes the energy of each atom into the energy of each edge formed by the atom and one of its local neighbors:
E_i=Σ(j∈neighbor(i))E_{ij}
Therefore, the output of the Allegro network is E_{ij}, which can be used to calculate the total energy of the system.
2. Result
1. In terms of prediction accuracy, Allegro performs similarly to NequIP on the revMD-17 dataset and achieves the best result on the QM9 dataset.
2. Based on the strictly local nature of Allegro, the author also demonstrates its performance advantages in parallel computing.
3. Thoughts
Related operations (such as irrep and tensor product) based on the group representation theory have high computation costs. Improving computing performance is an important topic of geometric GNNs. Traditional geometric GNNs based on message passing are inefficient in terms of parallel computing. Allegro does not rely on message passing. This assures strict locality while making excellent use of parallel computing. Now, MindSpore has started integrating its advantages into Allegro. The MindSpore implementation of Allegro will be added to MindSpore Chemistry soon. In addition, when designing a model to be applied on large molecular systems, a developer should consider the parallel computing efficiency in the first place.
References
[1]Keith T Butler, Daniel W Davies, Hugh Cartwright, Olexandr Isayev, and Aron Walsh. 2018. Machine learning for molecular and materials science. Nature 559, 7715 (2018), 547–555.
[2]Tian Xie and Jeffrey C Grossman. 2018. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Physical Review Letters 120, 14 (2018), 145301.
[3]Keqiang Yan, Yi Liu, Yuchao Lin, and Shuiwang Ji. 2022. Periodic Graph Transformers for Crystal Material Property Prediction. In The 36th Annual Conference on Neural Information Processing Systems. 15066–15080.