MindSpore-Based Model Surpasses Alphafold in Terms of Protein Structure Prediction
MindSpore-Based Model Surpasses Alphafold in Terms of Protein Structure Prediction
A MindSpore-based protein structure prediction model claimed top spot in the Continuous Automated Model EvaluatiOn (CAMEO) charts for three consecutive weeks and ranked in the top 2 for two consecutive months, signaling the strength of its cutting-edge performance. This prestige sets the Mindspore model apart from competitors, thanks to powerful capabilities created by Ascend AI software and hardware in the AI for Science field.

CAMEO, a competition jointly hosted by the Swiss Institute of Bioinformatics and the University of Basel in Switzerland, is one of the most important competitions in the protein structure prediction field, attracting distinguished developers in the biocomputing field from around the world.
Unlike other competitions like CASP, CAMEO requires contestants to predict the latest 20 protein structures. The scores and rankings of each contestant are updated every week.
As a community project, the event adopts quality assessment criteria established by the community for different accuracy requirements, covering coverage, local accuracy, and completeness.

Protein Data Bank (PDB) usually pre-releases sequence structures every Friday, and these are collected by CAMEO's team who preprocess and then submits them to registered servers for evaluation. The evaluation results are typically published the following Wednesday.
CAMEO currently supports three categories: protein structure modeling (3D), protein model quality assessment (QE), and structures and complexes (Beta 3D). Protein contact prediction (CP) and ligand binding (LB) have been discontinued.
CAMEO servers can be registered as public servers (the server name and data are available for public domain), or as development servers (server number: server_X_) that allow only developers to download data. For details about the registered server list, visit https://www.cameo3d.org/cameong_servers/.
CAMEO targets are evaluated against PDB's pre-released reference structures every week, and these structures are submitted to all registered servers. There are two types of targets that cannot be submitted for 3D protein structure prediction: those with templates covering ≥ 70% of the sequence and sequence identity is > 85%, or those with a sequence length > 250 and undiscovered amino acids are < 40.
All the submissions of monomeric and homo-oligomeric targets are assessed within a specified period, with inappropriate targets excluded after examination.
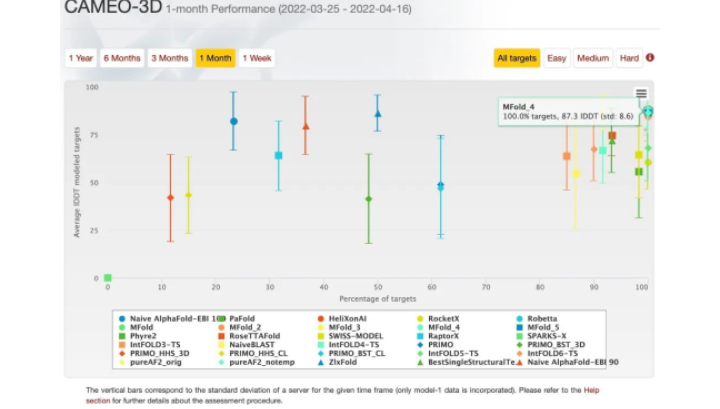
The difficulty of a target is determined through the evaluation of the average accuracy (lDDT) over the models received by servers, and lDDT-based targets are classified into three types: Easy (lDDT ≥ 75), Medium (lDDT: 50-75), Hard (lDDT < 50).
The fraction of atoms below a cutoff is similar to the percentage of Cα atoms in the predictions that deviate from the target by limited and specified distance (in Å) cut-off for different sequence-dependent superpositions. This method uses distance cut-off values of 0.5, 1, 2 and 4 Å, whereas superposition is calculated using the LGA program, a method for finding 3D similarities in protein structures.
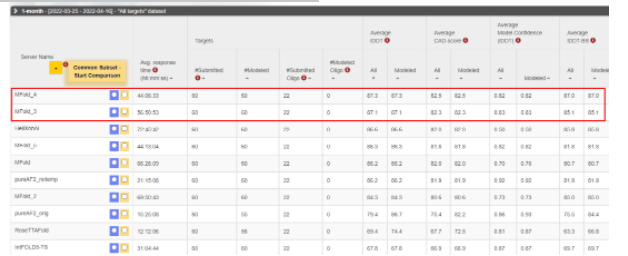
Under such a strict assessment system, the MindSpore-based model reaches over 85 points, greatly demonstrating its superb performance and capabilities.

The CAD-score, a contact area difference-based function, provides a unified framework for evaluating single-domain, multi-domain, and even multisubunit protein structure models varying in accuracy and integrity. Providing a highly-correlated base with GDT-TS on single-domain structures, the CAD-score emphasizes the physical realism of superposition-free models. Performance values of CAD-score ranges from 0 to 100 (0 the worst, and 100 the best).
A model's coverage refers to the percentage of residue in the model, where structure information is of great use in the experimental structure with respect to the length of the submitted sequence. Some CAMEO targets may have low coverage, and will be removed in future projects.
The GDT_HA (Global Distance Test) score identifies sets of residuals in different sequence-dependent superposition predictions that deviate from the target by no more than a specified Cα distance. This method uses distance cut-off values of 0.5, 1, 2 and 4 Å.
The GDC score identifies sets of residuals in different sequence-dependent superposition predictions that deviate from the target by no more than a specified Cα distance. This method uses distance cut-off values of 0.5, 1, 2 and 4 Å, and the GDC score is calculated with LGA (version 2009/5).
The lDDT score (local Distance Difference Test on All Atoms) evaluates the quality of the local atomic environment of a model. lDDT scores the correctly predicted inter-atomic distances in a model at different thresholds, but does not depend on a global superposition of the prediction and target structure.
Specifically, interaction distances (cutoff 15 Å) between atoms in the reference protein structure are compared with distances between corresponding atoms in the predictions. If the difference between the two distances is within a defined threshold, the interaction will be preserved. The final lDDT-all score is computed by averaging the fraction of correctly modeled interactions for the following four distance difference thresholds: 0.5, 1, 2, and 4 Å (the same thresholds as GDT_HA). A filter based on the Engh and Huber bond lengths and angles removes stereochemical violations and steric clashes. CAMEO additionally offers a Cα - based lDDT score.
The lDDT-BS score (local Distance Difference Test - Binding Site) measures the accuracy of residue which form the binding site(s) on the target structure. Here the binding site is defined as the set of amino acid residue in the reference protein structure that have at least one atom within a 4.0 Å radius of any atom of the ligand (3.0 Å for ions). Only ligands that form non-covalent interactions with the target are considered. Common solvent molecules are also excluded from the analysis based on a blacklist approach. The lDDT score is calculated for contacts within the binding site only, with a custom inclusion radius Ro of 10 Å and the standard thresholds of 0.5, 1, 2, and 4 Å.
When the reference structure contains several biologically relevant ligands, the lDDT-BS score is the average of the lDDT-BS scores of each binding site. This score is only calculated for targets where the experimental structure incorporates a ligand. When a binding site is at the interface of a homo-oligomeric structure and the prediction does not show the same oligomeric state, all possible chain mappings are assessed and the highest-scoring one is retained. Targets which cover only part of a hetero-oligomeric complex are not evaluated, since the modeled form might differ from its natural state and among evolutionarily related hetero-oligomeric complexes.
The evaluation of Model Confidence lDDT Values assesses the error estimates given as deviation in Å with a ROC AUC analysis. A residue is classified as correctly modeled if its local lDDT value is higher than or equal to 0.6. The local scores shown on the individual model pages are a measure of how well the model fits to the reference structure at a given residue position. Local scores are extracted from the output when calculating the average accuracy. Note that the ROC AUC is not defined for models with better or worse lDDT values (greater or smaller than 0.6).

In November 2021, Huawei jointly launched a tool for protein structure prediction and inference that runs on AlphaFold2 algorithms. In February 2022, Huawei, along with teams from the China Changping Laboratory, Peking University's Biomedical Pioneering Innovation Centre (BIOPIC) and Chemistry and Molecular Engineering College, and Shenzhen Bay Laboratory, released the model that streamlines training processes and improves efficiency by two to three times.
Running on the Ascend AI software and hardware platform, the tool enables mixed precision, slashes single-step iteration from 20 seconds to 12 seconds, and supercharges performance by over 60%. In addition, based on MindSpore's memory overcommitment feature, its training sequence length is increased from 384 to 512.
The record-breaking prediction result and rank in CAMEO signals a milestone for MindSpore in protein structure prediction, filling a scientific gap in the domestic field. Despite achieving a training accuracy that is very close to AlphaFold2, the MindSpore team doesn't stop innovating, and continually improves its algorithms, training scales, and software and hardware compatibilities to lead the industry.
Looking forward, MindSpore hopes to cooperate with more academic and industry partners to continuously make breakthroughs in protein structure prediction and fuel pharmaceutical development.
To learn more about MindSpore and the protein folding code, visit the MindSpore official website and the code repository at Gitee.