MindSpore Chemistry文档
介绍
传统化学研究长期以来面临着众多挑战,实验设计、合成、表征和分析的过程往往耗时、昂贵,并且高度依赖专家经验。AI与化学的协同可以克服传统方法的局限性、开拓全新的研究范式,结合AI模型与化学知识,可以高效处理大量数据、挖掘隐藏的关联信息,构建仿真模型,从而加快化学反应的设计和优化,实现材料的性质预测,并辅助设计新材料。
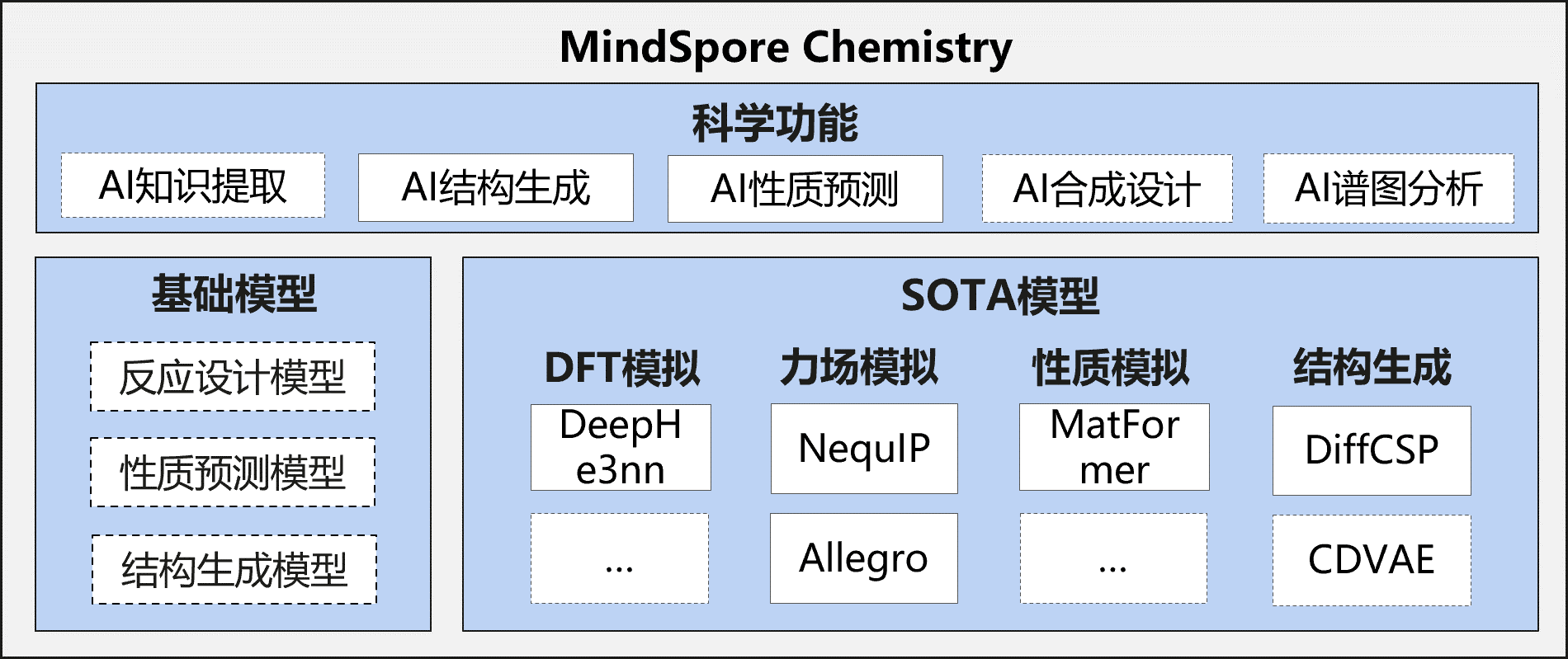
MindSpore Chemistry(MindChemistry)是基于MindSpore构建的化学领域套件,支持多体系(有机/无机/复合材料化学)、多尺度任务(微观分子生成/预测、宏观反应优化)的AI+化学仿真,致力于高效使能AI与化学的融合研究,践行和牵引AI与化学联合多研究范式跃迁,为化学领域专家的研究提供全新视角与高效的工具。
最新消息
2025.03.30 MindChemistry 0.2.0版本发布,包括多个应用案例,支持NequIP、Allegro、DeephE3nn、Matformer以及DiffCSP模型。
2024.07.30 MindChemistry 0.1.0版本发布。
特性
应用案例
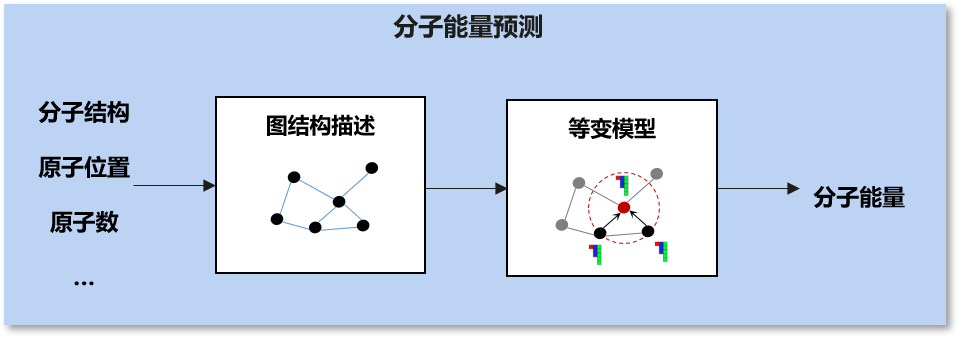
力场模拟:
体系:有机化学
数据:Revised Molecular Dynamics 17(rMD17)数据集。rMD17数据集包含了多种有机化合物的分子动力学性质,提供化合物的原子位置、原子数等描述信息以及能量、力场等性质信息。
任务:分子能量预测。我们集成了NequIP模型[2]、Allegro模型[3],根据分子体系中各原子的位置与原子数信息构建图结构描述,基于等变计算与图神经网络,计算出分子体系能量。
DFT模拟:
体系:材料化学
数据:双层石墨烯数据集。该数据集包含了原子位置、原子数等描述信息以及哈密顿量等性质信息。
任务:密度泛函理论哈密顿量预测。我们集成了DeephE3nn模型[4],基于E3的等变神经网络,利用原子的结构去预测其的哈密顿量。
性质预测:
体系:材料化学
数据:JARVIS-DFT 3D数据集。该数据集包含了晶体材料的原子位置、原子数等描述信息以及能量、力场等性质信息。
任务:晶体材料性质预测。我们集成了Matformer模型[5],基于图神经网络和Transformer架构的模型,用于预测晶体材料的各种性质。
结构生成:
体系:材料化学
数据:
Perov-5:钙钛矿数据集,每个晶胞中固定5个原子,结构接近。
Carbon-24:碳晶体数据集,包含6到24个碳原子的不同结构。
MP-20:MP数据集中的实验数据,胞内不超过20个原子。
MPTS-52:MP-20的进阶版,胞内最多52个原子。
任务:晶体材料结构预测。集成了 DiffCSP 模型[5],基于图神经网络和扩散模型,预测晶体材料的结构。
安装教程
版本依赖关系
由于MindChemistry与MindSpore有依赖关系,请根据下表中所指示的对应关系,在MindSpore下载页面下载并安装对应的whl包。
MindChemistry |
分支 |
MindSpore |
Python |
|---|---|---|---|
master |
master |
>=2.3 |
>=3.8 |
0.2.0 |
r0.7 |
>=2.5.0 |
>=3.11 |
0.1.0 |
r0.6 |
>=2.2.12 |
>=3.8 |
依赖安装
pip install -r requirements.txt
硬件支持情况
硬件平台 |
操作系统 |
状态 |
|---|---|---|
Atlas A2训练系列产品 |
Ubuntu-x86 |
✔️ |
Ubuntu-aarch64 |
✔️ |
|
EulerOS-aarch64 |
✔️ |
|
CentOS-x86 |
✔️ |
|
CentOS-aarch64 |
✔️ |
源码安装
从Gitee下载源码
git clone https://gitee.com/mindspore/mindscience.git cd {PATH}/mindscience/MindChemistry
编译昇腾Ascend后端源码
bash build.sh -e ascend
安装编译所得whl包
cd {PATH}/mindscience/MindChemistry/output pip install mindchemistry_*.whl
社区
核心贡献者
感谢以下开发者做出的贡献:
wujian, wangyuheng, Lin Peijia, gengchenhua, caowenbin, Siyu Yang
贡献指南
如何贡献您的代码,请点击此处查看:贡献指南
许可证
引用
[1] Batzner S, Musaelian A, Sun L, et al. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials[J]. Nature communications, 2022, 13(1): 2453.
[2] Musaelian A, Batzner S, Johansson A, et al. Learning local equivariant representations for large-scale atomistic dynamics[J]. Nature communications, 2023, 14(1): 579.
[3] Xiaoxun Gong, He Li, Nianlong Zou, et al. General framework for E(3)-equivariant neural network representation of density functional theory Hamiltonian[J]. Nature communications, 2023, 14: 2848.
[4] Keqiang Yan, Yi Liu, Yuchao Lin, Shuiwang ji, et al. Periodic Graph Transformers for Crystal Material Property Prediction[J]. arXiv:2209.11807v1 [cs.LG] 23 sep 2022.
[5] Jiao Rui and Huang Wenbing and Lin Peijia, et al. Crystal structure prediction by joint equivariant diffusion[J]. Advances in Neural Information Processing Systems, 2024, 36.