MindSpore Chemistry
Introduction
Conventional chemistry studies have long been confronted with numerous challenges. The process of experimental design, synthesis, characterization, and analysis can be time-consuming, costly, and highly dependent on experts’ experiences. The synergy between AI and chemistry offers unprecedented opportunities to overcome the limitations of conventional approaches and unlock new frontiers in scientific discovery and innovation. AI techniques can efficiently process vast amount of data, mining underneath patterns and generating predictive models. By leveraging AI, chemistry and material science researchers can accelerate the design and optimization of chemical processes and the design and analysis of novel materials.
MindSpore Chemistry(MindChemistry) is a toolkit built on MindSpore endeavoring to integrate AI with conventional chemistry research. It supports multi-scale tasks including molecular generation, property prediction and synthesis optimization on multiple chemistry systems such as organic, inorganic and composites chemistry systems. MindChemistry dedicates to enabling the joint research of AI and chemistry with high efficiency, and seek to facilitate an innovative paradigm of joint research between AI and chemistry, providing experts with novel perspectives and efficient tools.
Latest News
2025.03.30 MindChemistry 0.2.0 has been released, featuring several powerful applications, including NequIP, Allegro, DeephE3nn, Matformer, and DiffCSP.
2024.07.30 MindChemistry 0.1.0 has been released.
Features
Applications
Force Prediction:
Scenario: Organic chemistry
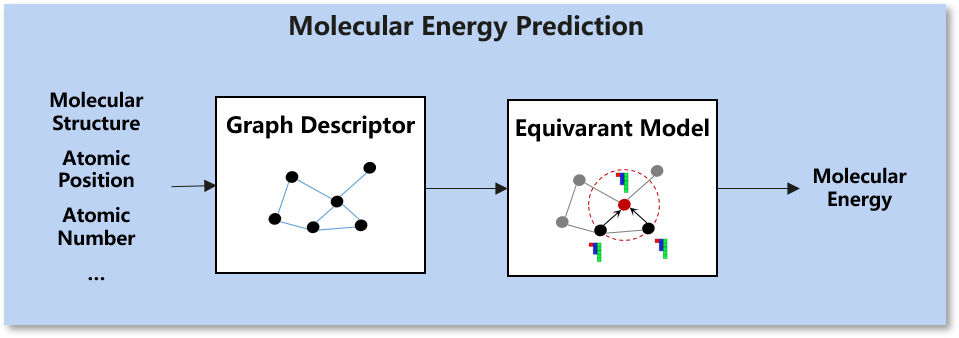
Dataset: Revised Molecular Dynamics 17(rMD17). rMD17 dataset includes molecular dynamics simulations of multiple organic chemical moleculars. It provides chemical desciptive information such as the atomic numbers and positions as well as molecular property information such as energies and forces.
Task: Molecular energy prediction. We integrate the NequIP model [1] and Allegro model [2], according to the position of each atom in the molecular system and structure description of the atomic number information construction diagram, and calculate the energy of the molecular system based on the equivariant calculation and graph neural network.
DFT Prediction:
Scenario: Materials Chemistry
Dataset: Bilayer graphene dataset. The dataset contains descriptive information such as atomic positions and atomic numbers, as well as property information such as Hamiltonian.
Task: Density Functional Theory Hamiltonian Prediction. We integrate the DeephE3nn model [3], an equivariant neural network based on E3, to predict a Hamiltonian by using the structure of atoms.
Property Prediction:
Scenario: Materials Chemistry
Dataset: JARVIS-DFT 3D dataset. The dataset contains descriptive information such as atomic position and atomic number of crystal materials, as well as property information such as energy and force field.
Task: Prediction of crystalline material properties. We integrate the Matformer model [4] based on graph neural networks and Transformer architectures, for predicting various properties of crystalline materials.
Structure Generation:
Scenario: Materials Chemistry
Dataset:
Perov-5: A perovskite dataset in which each unit cell contains five fixed atoms, and the structures are relatively similar.
Carbon-24: A carbon crystal dataset, where each crystal contains between 6 and 24 carbon atoms, with various different material structures.
MP-20: A dataset collected from the MP database, featuring experimental structures with up to 20 atoms per unit cell. The materials and structures are highly diverse.
MPTS-52: An advanced version of MP-20, expanding the number of atoms per unit cell to 52. The materials and structures are highly diverse.
Task: Crystal material structure prediction. We integrated the DiffCSP model [5], which is based on a graph neural network and diffusion model architecture, to predict the crystal material structures given their composition.
Installation
Version Dependency
Because MindChemistry is dependent on MindSpore, please click MindSpore Download Page according to the corresponding relationship indicated in the following table. Download and install the corresponding whl package.
MindChemistry |
Branch |
MindSpore |
Python |
|---|---|---|---|
master |
master |
>=2.3 |
>=3.8 |
0.2.0 |
r0.7 |
>=2.5.0 |
>=3.11 |
0.1.0 |
r0.6 |
>=2.2.12 |
>=3.8 |
Dependency
pip install -r requirements.txt
Hardware
Hardware |
os |
Status |
|---|---|---|
AtlasA2 training series |
Ubuntu-x86 |
✔️ |
Ubuntu-aarch64 |
✔️ |
|
EulerOS-aarch64 |
✔️ |
|
CentOS-x86 |
✔️ |
|
CentOS-aarch64 |
✔️ |
source code install
Download source code from Gitee
git clone https://gitee.com/mindspore/mindscience.git cd {PATH}/mindscience/MindChemistry
Compile in Ascend backend
bash build.sh -e ascend -j8
Install whl package
cd {PATH}/mindscience/MindChemistry/output pip install mindchemistry_*.whl
Community
Core Contributor
Thanks goes to these wonderful people:
wujian, wangyuheng, Lin Peijia, gengchenhua, caowenbin, Siyu Yang
Contribution Guide
Please click here to see how to contribute your code: Contribution Guide
License
References
[1] Batzner S, Musaelian A, Sun L, et al. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials[J]. Nature communications, 2022, 13(1): 2453.
[2] Musaelian A, Batzner S, Johansson A, et al. Learning local equivariant representations for large-scale atomistic dynamics[J]. Nature communications, 2023, 14(1): 579.
[3] Xiaoxun Gong, He Li, Nianlong Zou, et al. General framework for E(3)-equivariant neural network representation of density functional theory Hamiltonian[J]. Nature communications, 2023, 14: 2848.
[4] Keqiang Yan, Yi Liu, Yuchao Lin, Shuiwang ji, et al. Periodic Graph Transformers for Crystal Material Property Prediction[J]. arXiv:2209.11807v1 [cs.LG] 23 sep 2022.
[5] Jiao Rui and Huang Wenbing and Lin Peijia, et al. Crystal structure prediction by joint equivariant diffusion[J]. Advances in Neural Information Processing Systems, 2024, 36.